Research
Function-valued trait evolution
Phylogenetic comparative methods, such as ancestral state reconstruction and tests of correlated trait evolution, typically assume fixed trait value means for each species. In the case of phenotypically plastic traits, developmental trajectories, environmental variation, and exposure to toxic substances, this assumption is violated, as traits are better represented as non-linear mathematical functions. In Goolsby (2015, Syst Biol), I propose methods for studying function-valued trait evolution in a phylogenetic context, which extend phylogenetic comparative methods such as ancestral state reconstruction and phylogenetics signal tests to a function-valued PGLS framework.
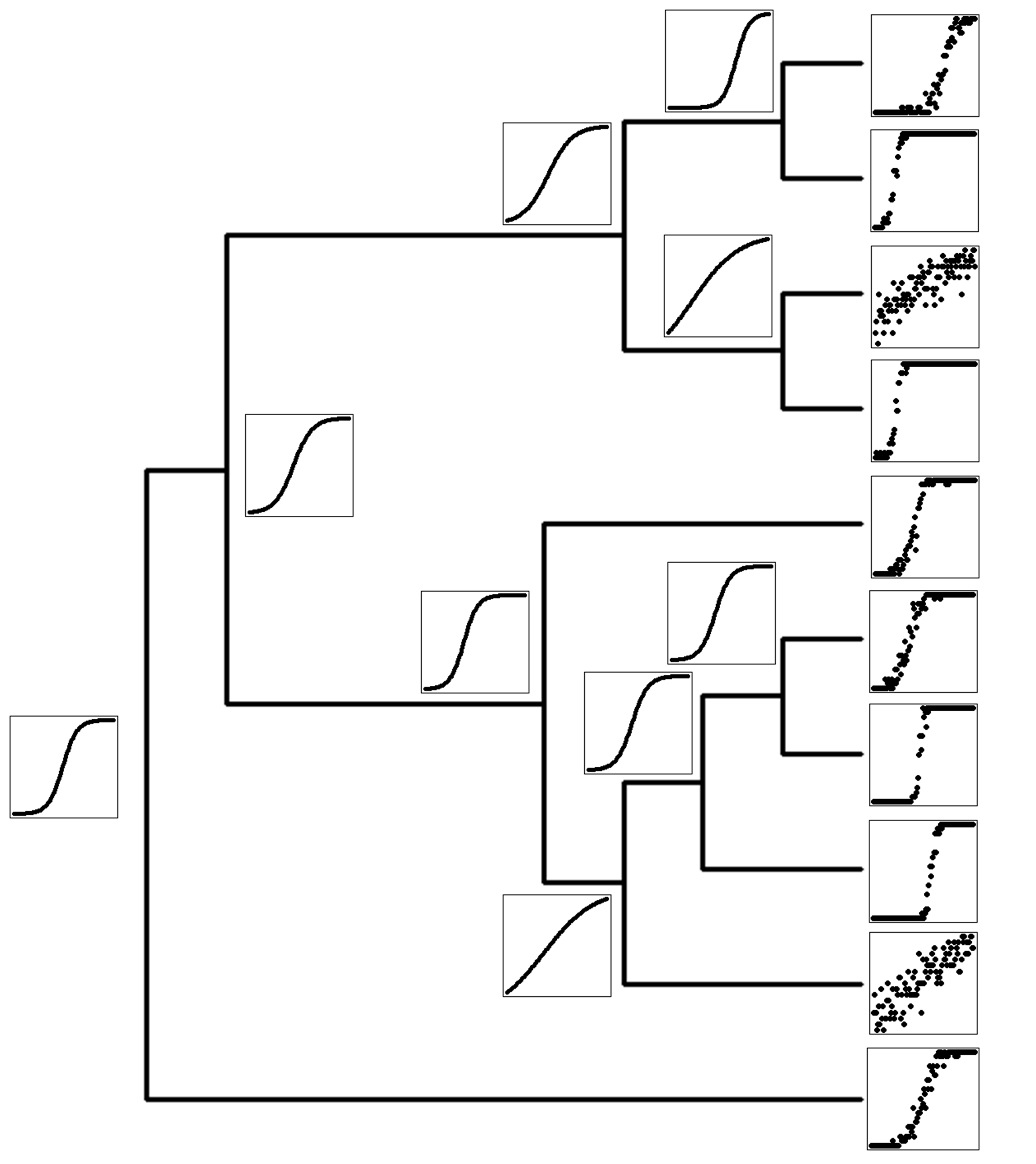
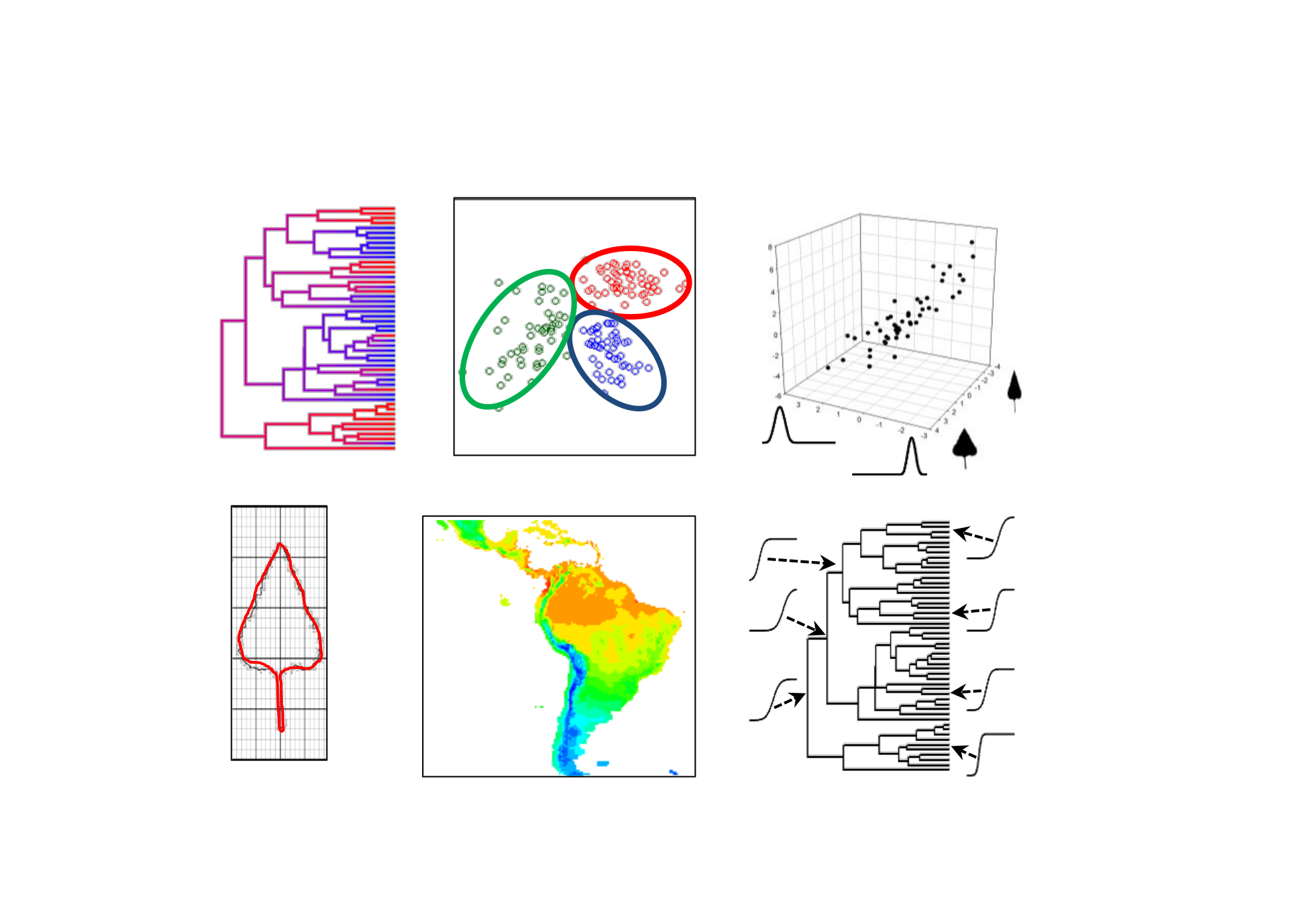
High-dimensional trait evolution
Function-valued traits and, more generally, high-dimensional triats, such as gene expression levels, morphometric data, and multifaceted physiological suites/syndromes, cannot be approached with typical multivariate statstics, as the number of trait dimensions typically far exceeds the number of observations (species). In Goolsby (2016, Syst Biol), I propose a parametric bootstrapping approach based on maximum pseudolikelihood parameter estimation, which allows for high-dimensional traits to be studied in phylogenetic comparative context. Unlike distance-based phylogenetic comparative methods (Adams 2014-2016), which are limited to Brownian motion evolution and suffer from issues related to statistical power, the approach described here allows for flexbile model specification including alternative (non-Brownian) evolutionary models in a statistically powerful framework. These methods are also implemented in the R package phylocurve.
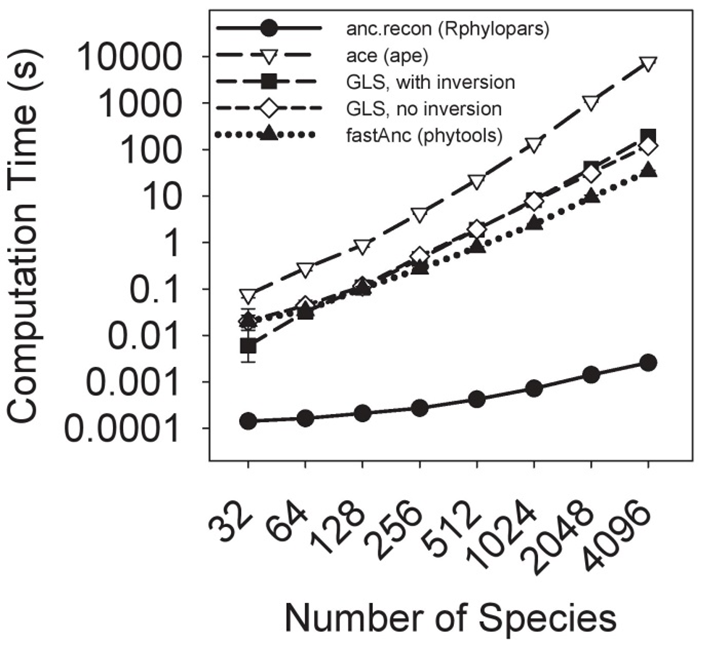
Fast phylogenetic comparative methdos
Fast comparative methods are critical to keeping up with the ever-increasing size of phylogenetic trees in studies, as well as for statistical methods requiring thousands or millions of repeated calculations. In Goolsby, Bruggeman, and Ané (2016, Methods in Ecol & Evol) and in the R package Rphylopars, we develop fast multivaraite methods for analyzing extremely large multivariate datasets with complex issues such as within-species variation, missing data, and non-neutral evolutionary models, which perform several thousand times faster than previous methods, thus allowing extremely large and previously infeasible problems to be computed in seconds or minutes (rather than hours or days). I have also developed an algorithm for maximum likelihood ancestral character reconstruction which performs several orders of magnitude faster than the next-fastest available method (currently in review, implemented in the Rphylopars function anc.recon).
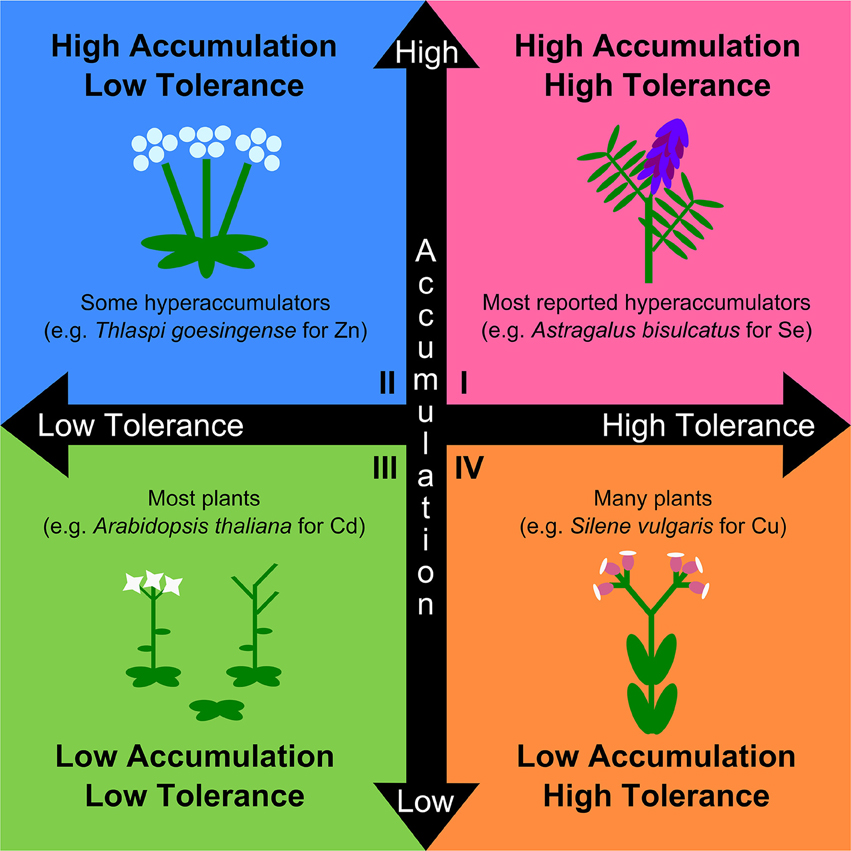
Heavy metal hyperaccumulation
More than 500 plant species have demonstrated the ability to absorb metals or metalloids from soils and concentrate them at extremely high levels in leaf tissues. This phenomenon, known as hyperaccumulation, has profound ecological implications including herbivory deterrence, trophic transfer of metals, and modifications of soil chemistry. Although extensively studied, few studies have examined the evolutionary trajectoies of heavy metal tolerance hyperaccumulation at the genus level. In Goolsby and Mason (2015, Frontiers in Plant Sci), I argue that metal tolerance and accumulation (two putatively separate traits) should be studied as distinct physiological and evolutionary traits. I further argue in Goolsby and Mason (2016, Frontiers in Plant Sci) that experimental soil metal gradients provides a powerful function-valued approach to untangling the evolutionary dynamics of the separate traits of hyperaccumulation and tolerance. I am currently studying the correlated evolutionary history of metal tolerance and accumulation as separate function-valued traits in the multi-metal hyperaccumulator genus Helianthus (the sunflowers).